

Abstract
Human severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is most closely related, by average genetic distance, to two coronaviruses isolated from bats, RaTG13 and RmYN02. However, there is a segment of high amino acid similarity between human SARS-CoV-2 and a pangolin isolated strain, GD410721, in the receptor binding domain (RBD) of the spike protein, a pattern that can be caused by either recombination or by convergent amino acid evolution driven by natural selection. We perform a detailed analysis of the synonymous divergence, which is less likely to be affected by selection than amino acid divergence, between human SARS-CoV-2 and related strains. We show that the synonymous divergence between the bat derived viruses and SARS-CoV-2 is larger than between GD410721 and SARS-CoV-2 in the RBD, providing strong additional support for the recombination hypothesis. However, the synonymous divergence between pangolin strain and SARS-CoV-2 is also relatively high, which is not consistent with a recent recombination between them, instead it suggests a recombination into RaTG13. We also find a 14-fold increase in the dN/dS ratio from the lineage leading to SARS-CoV-2 to the strains of the current pandemic, suggesting that the vast majority of non-synonymous mutations currently segregating within the human strains have a negative impact on viral fitness. Finally, we estimate that the time to the most recent common ancestor of SARS-CoV-2 and RaTG13 or RmYN02 based on synonymous divergence, is 51.71 years (95% C.I., 28.11-75.31) and 37.02 years (95% C.I., 18.19-55.85), respectively.
Introduction
The Covid19 pandemic is perhaps the biggest public health and economic threat that the world has faced for decades (Li, et al. 2020; Wu, et al. 2020; Zhou, Yang, et al. 2020). It is caused by a coronavirus (Lu, et al. 2020; Zhang and Holmes 2020), Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), an RNA virus with a 29,891 bp genome consisting of four major structural genes (Wu, et al. 2020; Zhou, Yang, et al. 2020). Of particular relevance to this study is the spike protein which is responsible for binding to the primary receptor for the virus, angiotensin-converting enzyme 2 (ACE2) (Wan, et al. 2020; Wu, et al. 2020; Zhou, Yang, et al. 2020).
Human SARS-CoV-2 is related to a coronavirus (RaTG13) isolated from the bat Rhinolophus affinis from Yunnan province of China (Zhou, Yang, et al. 2020). RaTG13 and the human strain reference sequence (Genbank accession number MN996532) are 96.2% identical and it was first argued that, throughout the genome, RaTG13 is the closest relative to human SARS-CoV-2 (Zhou, et al. 2020). Zhang, et al. 2020 showed that RaTG13 and SARS-CoV-2 were 91.02% and 90.55% identical, respectively, to coronaviruses isolated from pangolins (Pangolin-CoV), which therefore form a close outgroup to the SARS-CoV-2+RaTG13 clade. Furthermore, five key amino acids in the receptor-binding domain (RBD) of spike were identical between SARS-CoV-2 and Pangolin-CoV, but differed between those two strains and RaTG13. (Lam, et al. 2020) independently made similar observations and additionally showed that when analyzing a window of length 582bp in the RBD, nonsynonymous mutations support a phylogenetic tree with SARS-CoV-2 and Pangolin-CoV as sister-groups, while synonymous mutations do not. They discuss two possible explanations for their results, one which includes recombination and another which includes selection-driven convergent evolution. Boni, et al. 2020 found little evidence of recombination between SARS-CoV-2 and Pangolin-CoV using the recombination analysis software 3SEQ (Lam, et al. 2018), but argued that if there has been recombination, it likely occurred into RaTG13 from an unknown divergent source. This would explain the amino acid similarity between SARS-CoV-2 and Pangolin-CoV in the RBD as an ancestral trait that has been lost (by recombination) in RaTG13. Using a phylogenetic analysis they also dated the RaTG13 and SARS-CoV-2 divergence to be between 40 to 70 years.
Recently, Zhou et al. (2020) discovered a viral strain (Zhou, et al. 2020), RmYN02 from the bat Rhinolophus malayanus, with a reported 97.2% identity in the ORF1ab gene but with only 61.3% sequence similarity to SARS-CoV-2 in the RBD. Moreover, the RmYN02 strain also harbors multiple amino acid insertions at the functional polybasic (furin) cleavage site (Zhou, et al. 2020) that was thought to be unique to the human SARS-CoV-2 (Andersen, et al. 2020).
To analyze the history of these sequences further, we here focus on patterns of synonymous divergence, which has received less focus, but also is less likely to be affected by selection than amino acid divergence. We develop a bias corrected estimator of synonymous divergence specific for SARS-CoV-2 and related strains, and analyze divergence using both sliding windows and a whole-genome approach between SARS-CoV-2 and related viral strains.
Materials and methods
Sequence data
The pangolin virus sequences, GD410721 and GX_P1E, were downloaded from GISAID with accession numbers EPI_ISL_410721 and EPI_ISL_410539, respectively, and RmYN02 was provided by E. C. Holmes. All other sequences analyzed in this study were downloaded from either NCBI Genbank or National Microbiology Data Cente (NMDC). The accession codes for non-human sequences can be found in Supplementary Table 2 and the accession codes for human sequences can be found in Supplementary Table 3.
BLAST searches
Sequences for blast databases were downloaded on March 26, 2020 from the following sources: EMBL nucleotide libraries for virus (ftp://ftp.ebi.ac.uk/pub/databases/embl/release/std), NCBI Virus Genomes (ftp://ftp.ncbi.nlm.nih.gov/genomes/Viruses), NCBI Virus Genbank Entries (ftp://ftp.ncbi.nlm.nih.gov/genomes/genbank/viral/), NCBI Influenza Genomes (ftp://ftp.ncbi.nlm.nih.gov/genomes/INFLUENZA/), all Whole Genome Shotgun (https://www.ncbi.nlm.nih.gov/genbank/wgs/) assemblies under taxonomy ID 10239, along with GISAID Epiflu and EpiCoV databases. Sequences from Valitutto et al. (2020) were also added to the database. Blast databases were created using the default parameters for makeblastdb. Blast searches were performed using blastn with parameters “-word_size 7 -reward 1 -penalty - 3” and all other parameters as the default settings. All the blast hits to different Guangdong pangolin viral strain sequences were merged as one hit, and the blast hits to different Guangxi pangolin viral strain sequences were also merged.
Alignment
To obtain an in-frame alignment of the genomes, we first identified the coding sequences of each viral strain using independent pairwise alignments with the coding sequences of the SARS-CoV-2 (Wuhan-Hu-1) genome. The alignments were performed using MAFFT (v7.450) (Katoh and Standley 2013) with parameters “--maxiterate 1000 --localpair”. We then performed multiple sequence alignments for the coding sequences of each gene using PRANK (Loytynoja 2014) (v.170427) with parameters “-codon -F”. Finally, the alignments for all genes were concatenated following their genomic order. ORF1a was excluded without loss of any nucleotides, since its sequence is a subset of ORF1ab.
Recombination detection
We detected possible recombination events across the genome using the 3SEQ algorithm (Lam, et al. 2018) implemented in RDP (Martin, et al. 2015) (version Beta 5.5). The analysis was performed on the multiple sequence alignment consisting of the five viral strains with the ‘automatied 3Seq’ mode in RDP. All regions in which any of the viral strains showed evidence (p < 0.05) of recombination (Supplementary Table 5) were removed in subsequent analyses from all strains when stating that recombination regions were removed.
Tree estimation
We estimated phylogenetic trees using two methods: Neighbor Joining (NJ) and Maximum Likelihood (ML). The NJ trees were estimated using dN or dS distance matrices which estimated using codeml (Yang 2007) with parameters “runmode= −2, CodonFreq = 2, cleandata = 1”. To obtain bootstrap values, we bootstrapped the multiple sequence alignments 1,000 times, repeating the inference procedure for each bootstrap sample. The NJ tree was estimated using the ‘neighbor’ software from the PHYLIP package (Felsenstein 2009). For ML trees, we used IQ-TREE (Nguyen, et al. 2015) (v1.5.2) with parameter “-TEST -alrt 1000” which did substitution model selection for the alignments and performed maximum-likelihood tree estimation with the selected substitution model for 1,000 bootstrap replicates. For this analysis, we masked all regions (Supplementary Table 5) identified by 3SEQ (Lam, et al. 2018) to be recombinant in any of the five studied viral genome. The coordinates (based on the Wuhan-Hu-1 genome) of the three recombination regions (merged set of all the regions in Supplementary Table 5) were: 14611-15225, 21225-24252 and 25965-28297. We also estimate genome-wide divergence between RaTG13 and Wuhan-Hu-1 only excluding the region (position 22853-23092) where potential recombination was detected for the Wuhan-Hu-1 strain (Supplementary Table 5).
Simulations
We simulated divergence with realistic parameters for SARS-CoV-2 using a continuous time Markov chain under the F3×4 codon-based model (Goldman and Yang 1994; Muse and Gaut 1994; Yang, et al. 2000), which predicts codon frequencies from the empirical nucleotide frequencies in all 3 codon positions and using the global genomic maximum likelihood estimates of the transition/transversion bias κ(=2.9024) and the dN/dS ratio ω (=0.0392) estimated from the human SARS-CoV-2 comparison to the nearest outgroup sequence, RaTG13 (see Results). For the simulations of short 300 bp sequences we kept constant but varied time such that the number of synynoymous substitutions per synonymous sites, dS varied between 0.25 and 3.00. Estimates of dS > 3 are truncated to 3. For simulations of genome-wide divergence between RaTG13 and human strains, we fix dS at 0.1609 (the maximum likelihood estimate outside the RBD region reported in the Results section). In all cases, we use 10,000 independent replicate simulations for each parameter setting.
Estimation of sequence divergence in 300-bp windows
dN and dS were estimated using two different methods implemented in the PAML package (Yang 2007) (version 4.9d): a count-based method, YN00 (Yang and Nielsen 2000) as implemented in the program ‘yn00’ with parameters “icode = 0, weighting = 0, commonf3×4 = 0”, and a maximum-likelihood method (Goldman and Yang 1994; Muse and Gaut 1994) implemented in codeml applied with arguments “runmode= −2, CodonFreq = 2”. The estimates in 300-bp windows were further bias-corrected as described below.
Bias correction for dS estimates in 300-bp window
To correct for the biases observed in the estimation of dS (see results section) we identifed a quartic function which maps from  the estimates of d, into
the estimates of d, into  , the bias corrected estimate such that to a close approximation,
, the bias corrected estimate such that to a close approximation,  . To identify the coefficients of this function we used 10,000 simulations as previously described, on a grid of dS values (0.25, 0.5, 0.75, …, 3.0). We then identified coefficients such that sum of
. To identify the coefficients of this function we used 10,000 simulations as previously described, on a grid of dS values (0.25, 0.5, 0.75, …, 3.0). We then identified coefficients such that sum of  is minimized over all simulation values.
is minimized over all simulation values.
Results
Database searches
To identify possible viral strains that may have contributed, by recombination, to the formation of human SARS-CoV-2, we searched NCBI and EMBL virus entries along with GISAID Epiflu and EpiCov databases for similar sequences using BLAST in 100bp windows stepping every 10bp (Fig. 1B). The majority of the genome (78.1%, 2330/2982 of the windows) has one unique best hit, likely reflecting the high genetic diversity of the coronavirus. 21.9% of the genomic regions has multiple best hits, which suggests that these regions might be more conserved. Among the windows with unique best hits, 97.0% (2260/2330) of them were the RaTG13 or RmYN02 bat strains and 1.9% of them, including the ACE2 contact residues region of the S protein, were the pangolin SARS-CoV-2 virus. These observations are consistent with previous results that RaTG13 and RmYN02 are the most closely related viral strains, while the region containing the ACE2 contact residues is more closely related to the pangolin virus strains. A considerable amount of genomic regions (20 windows with unique hits) show highest sequence identity with other coronaviruses of the SARS-CoV-2 related lineage (Lam, et al. 2020) (bat-SL-CoVZC45 and bat-SL-CoVZXC21, (Hu, et al. 2018)). In addition, there were 6 windows whose unique top hits are coronavirus of a SARS-CoV related lineage (Lam, et al. 2020) (Supplementary Table 4). The mosaic pattern that different regions of the genome show highest identity to different virus strains is likely to have been caused by the rich recombination history of the SARS-CoV-2 lineage (Boni, et al. 2020; Patiño-Galindo, et al. 2020). Moreover, its unique connection with SARS-CoV related lineages in some genomic regions may suggest recombination between the ancestral lineage of SARS-CoV-2 and distantly related virus lineages, although more formal analyses are needed to determine the recombination history (see also Boni, et al. 2020 for further discussion). Searching databases with BLAST using the most closely related viral strains, RaTG13 and RmYN02, we observe a very similar pattern, as that observed for SARS-CoV-2, in terms of top hits across the genome (Fig. 1B), suggesting that these possible recombination events with distantly related lineages are not unique to the SARS-CoV-2 lineage, but happened on the ancestral lineage of both SARS-CoV-2, RaTG13, and RmYN02. A notable exception is a large region around the S gene, where RmYN02 show little similarity to both SARS-CoV-2 and RaTG13.
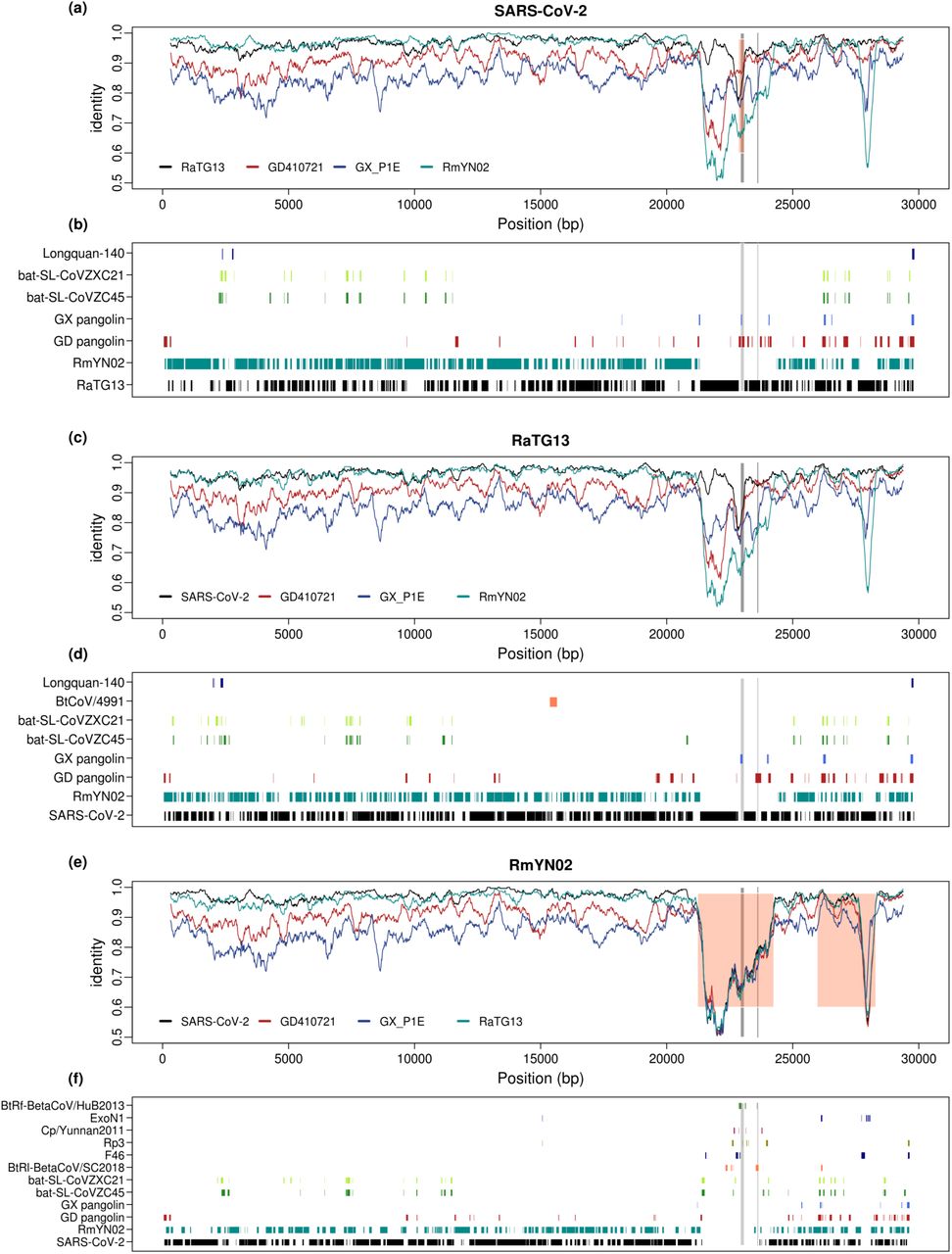
(a) 300 bp sliding-windows of nucleotide identity between SARS-CoV-2 and the four most closely related viral strains, RmYN02, RaTG13, GD410721 and GX_P1E. Orange shading marks the recombinant region in SARS-CoV-2 inferred by 3SEQ (details in Supplementary Table 5). (b) the plot lists all the viral strains that are the unique best BLAST hit in at least three 100-bp windows, when blasting with SARS-CoV-2, with the regions where each strain is the top blast hit marked. (b) and (c). Figures for RaTG13 (c, d) and RmYN02 (e, f) generated in the same way as for SARS-CoV-2 in (a) and (b). The ACE2 contact residues of RBD region (left) and the furin sites (right) of the S protein are marked in both plots with grey lines.
Sequence similarity
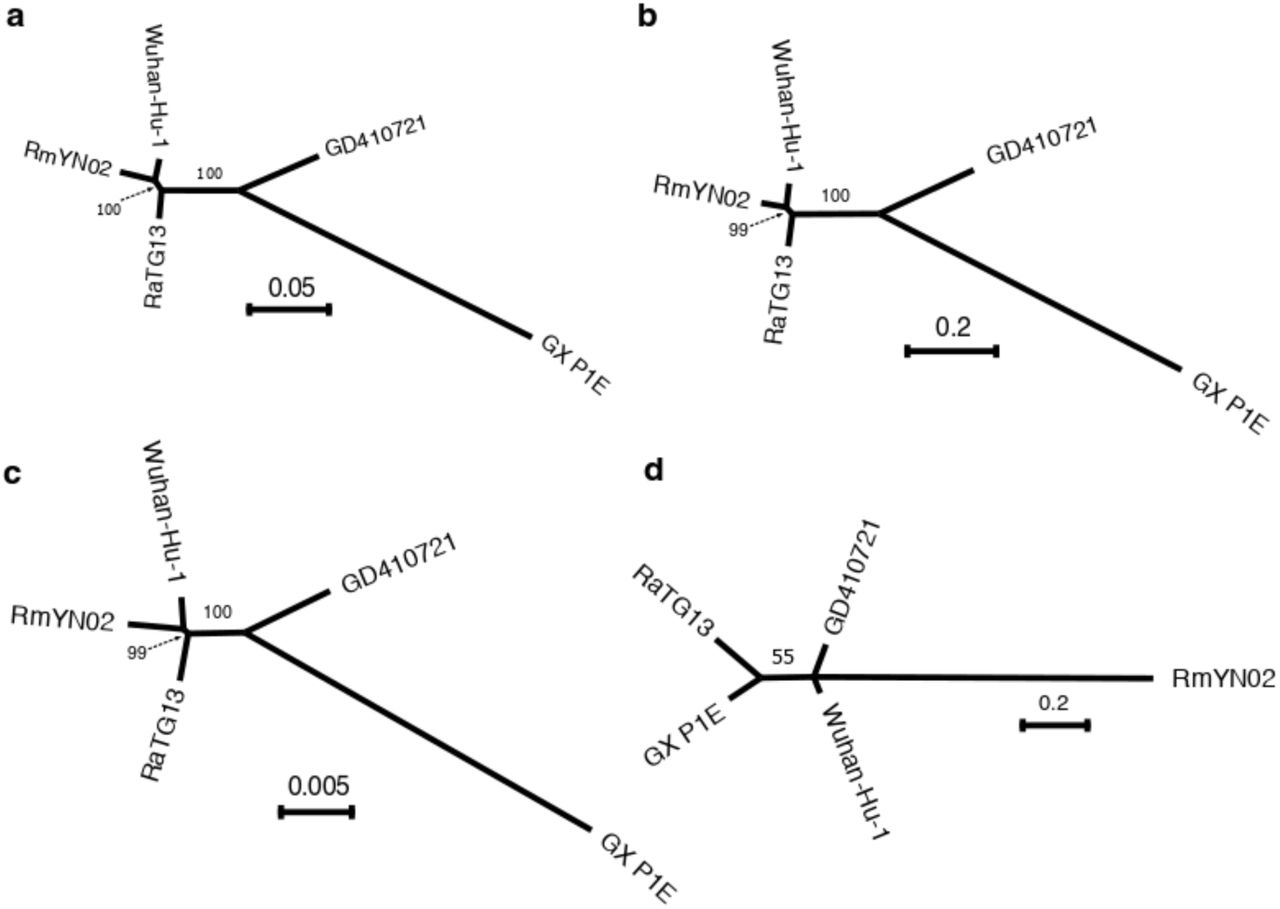
We focus further on analyzing SARS-CoV-2: Wuhan-Hu-1 as the human nCoV19 reference strain (Wu, et al. 2020) and the four viral strains with highest overall identity: the bat strains, RmYN02 and RaTG13 (Zhou, et al. 2020; Zhou, Yang, et al. 2020) and the Malayan pangolin strains, GD410721 and GX_P1E, which were isolated from samples seized by Guangdong and Guangxi Customs of China, respectively (Lam, et al. 2020; Xiao, et al. 2020). These four strains have previously been identified as the strains most closely related to SARS-CoV-2 (Lam, et al. 2020; Zhou, et al. 2020). The overall Neighbor-Joining and maximum-likelihood trees between the sequences closely related to human SARS-CoV-2 is shown in Fig. 2. RmYN02 has previously been reported to have disconcordant levels of divergence to SARS-CoV-2 in different parts of the genome, presumably as a result of recombination. To illustrate this, we performed recombination analyses with 3SEQ (Lam, et al. 2018) across the five viral genomes and identified several recombination regions affecting RmYN02 (Fig. 1C; Supplementary Table 5). Phylogenetic analysis (Fig. 2) in genome region with all recombination tracts (Supplementary Table 5) masked using Maximum-likelihood tree (Fig. 2A) and Neighbor-joining tree based on synonymous (Fig. 2B) or non-synoymous (Fig. 2C) mutation distance metrics consistently support RmYN02 as the nearest outgroup to human SARS-CoV-2, in contrast to previous analyses before the discovery of RmYN02, which instead found RaTG13 to be the nearest outgroup (Lam, et al. 2020; Zhou, Yang, et al. 2020). This observation is also consistent with the genome-wide phylogeny constructed in Zhou et al (Zhou, et al. 2020).

(a) Maximum-likelihood tree in genomic regions with recombination tracts removed; (b) Neighbor-joining tree using synonymous mutation (dS) distance in genomic regions with recombination tracts removed; (c) Neighbor-joining tree using non-synonymous mutation (dN) distances in genomic regions with recombination tracts removed; (d) The maximum-likelihoods tree at the receptor-binding domain ACE2 contact residues (51 amino acids) region. The bootstrap values are based on 1,000 replicates. The associated distance matrix for (b) and (c) can be found in Table 3.
We plot the overall sequence similarity (% nucleotides identical) between SARS-CoV-2 and the four other strains analyzed in windows of 300 bp (Fig. 1). Notice that the divergence between human SARS-CoV-2 and the bat viral sequences, RaTG13 and RmYN02, in most regions of the genome, is quite low compared to the other comparisons. A notable exception is the suspected recombination region in RmYN02 that has an unusual high level of divergence with all other viruses (Fig. 2E). However, there is also another exception: a narrow window in the RBD of the S gene where the divergence between SARS-CoV-2 and GD410721 is moderate and the divergences between GD410721 and both SARS-CoV-2 and RaTG13 are quite high and show very similar pattern. This would suggest a recombination event from a strain related to GD410721 into an ancestor of the human strain (Lam, et al. 2020; Xiao, et al. 2020; Zhang, et al. 2020), or alternatively, from some other species into RaTG13, as previously hypothesized (Boni, et al. 2020). We note that RmYN02 is not informative about the nature of this event as it harbors a long and divergent haplotype in this region, possibly associated with another independent recombination event with more distantly related viral strains (Fig. 2E). The other four sequences are all highly, and approximately equally, divergent from RmYN02 in this large region (Fig. 2E), suggesting that the RmYN02 strain obtained a divergent haplotype from the recombination event. When BLAST searching using 100-bp windows along the RmYN02 genome, we find no single viral genome as the top hit, instead the top hits are found sporadically in different viral strains of the SARS-CoV lineage (Fig. 2F), suggesting that the sequence of the most proximal donor is not represented in the database.
Estimating synonymous divergence and bias correction
While the overall divergence in the S gene encoding the spike protein could suggest the presence of recombination in the region, Lam et al. (2020) (Lam, et al. 2020) reported that the tree based on synonymous substitutions supported RaTG13 as the sister taxon to the human SARS-CoV-2 also in this region. That would suggest the similarity between GD410721 and human SARS-CoV-2 might be a consequence of convergent evolution, possibly because both strains adapted to the use of the same receptor. An objective of the current study is to examine if there are more narrow regions of the spike protein that might show evidence of recombination. We investigate this issue using estimates of synonymous divergence per synonymous site (dS) in sliding windows of 300 bp. However, estimation of dS is complicated by the high levels of divergence and extremely skewed nucleotide content in the 3rd position of the sequences (Table 1) which will cause a high degree of homoplasy. We, therefore, entertain methods for estimation that explicitly account for unequal nucleotide content and multiple hits in the same site such as maximum likelihood methods and the YN00 method (Yang and Nielsen 2000). Yang and Nielsen (2000) (Yang and Nielsen 2000) showed that for short sequences, some counting methods, such as the YN00 method, can perform better in terms of Mean Squared Error (MSE) for estimating dN and dS. However, it is unclear in the current case how best to estimate dS. For this reason, we performed a small simulations study (see Methods) for evaluating the performance of the maximum likelihood (ML) estimator of dN and dS (as implemented in codeml (Yang 2007)) under the F3×4 model and the YN00 method implemented in PAML. In general, we find that estimates under the YN00 are more biased with slightly higher MSE than the ML estimate for values in the most relevant regime of dS analyses of 300 bp regions < 1.5 (Fig. 3). However, we also notice that both estimators are biased under these conditions. For this reason, we perform a bias correction calibrated using simulations specific to the nucleotide frequencies and dN/dS ratio observed for SARS-CoV-2 (see Methods). The bias corrections we obtain are  , for the ML estimator and
, for the ML estimator and  for yn00. Notice that there is a trade-off between mean and variance (Fig. 3) so that the MSE becomes very large, particularly for the for yn00 method, after bias correction. For dS the estimates are generally not reliable, however, we note that for dS <1.5 the bias-corrected ML estimator tends overall to have slightly lower MSE, and we, therefore, use this estimator for analyses of 300 bp regions.
for yn00. Notice that there is a trade-off between mean and variance (Fig. 3) so that the MSE becomes very large, particularly for the for yn00 method, after bias correction. For dS the estimates are generally not reliable, however, we note that for dS <1.5 the bias-corrected ML estimator tends overall to have slightly lower MSE, and we, therefore, use this estimator for analyses of 300 bp regions.
Genome-wide nucleotide composition at the third position of the codons in the viral strains. The nucletodie compositions at the first and second positions can be found in Supplementary table 1.

(a) The mean of dS estimates using different methods; ML.corr and yn00.corr are the bias corrected versions of the ML and yn00 methods, respectively. (b) Errors in dS estimates as measured using the ratio of square root of mean squared error (MSE) to true dS. All the estimates are based on 10,000 simulations. ML: maximum-likelihood estimates using the f3×4 model in codeml; ML.corr, maximum-likelihood estimates with bias correction; yn00, count-based estimates in (Yang and Nielsen 2000); yn00.corr, yn00 estimates with bias correction. All dS estimates are truncated at 3, explaining the reduction in MSE with increasing values of dS as dS approaches 3.
Synonymous divergence
We estimate dN and dS under the F3×4 model in codeml (Goldman and Yang 1994; Muse and Gaut 1994) and find genome-wide estimates of dS = 0.1604, dN = 0.0065 (dN/dS = 0.0405) between SARS-CoV-2 and RaTG13 and 0.2043 (dN/dS = 0.1077) between SARS-CoV-2 and RmYN02. However, a substantial amount of this divergence might be caused by recombination with more divergent strains. We, therefore, infer recombination tracts using 3SEQ (Lam, et al. 2018) (Supplementary Table 5) and also estimate dN and dS for the regions with inferred recombination tracts removed from all sequences (Table 3). We then find values of dS = 0.1462 (95% C.I., 0.1340-0.1584) and dS = 0.1117 (95% C.I., 0.1019-0.1215) between SARS-CoV-2 and RaTG13 and RmYN02, respectively. This confirms that RmYN02 is the virus most closely related to SARS-CoV-2. The relative high synonymous divergence also shows that the apparent high nucleotide similarity between SARS-CoV-2 and the bat strains (96.2% (Zhou, Yang, et al. 2020) and 97.2% (Zhou, Chen, et al. 2020)) is caused by conservation at the amino acid level (dN/dS = 0.0410 and 0.0555) exacerbated by a high degree of synonymous homoplasy facilitated by a highly unusual nucleotide composition.
The synonymous divergence to the pangolin sequences GD410721 and GX_P1E in genomic regions with inferred recombination tracts removed is 0.5095 (95% C.I., 0.4794- 0.5396) and 1.0304 (95% C.I., 0.9669-1.0939), respectively. Values for other comparisons are shown in Tables 2 and 3. In comparisons between SARS-CoV-2 and more distantly related strains, dS will be larger than 1, and with this level of saturation, estimation of divergence is associated with high variance and may be highly dependent on the accuracy of the model assumptions. This makes phylogenetic analyses based on synonymous mutations unreliable when applied to these more divergent sequences. Nonetheless, the synonymous divergence levels seem generally quite compatible with a molecular clock with a dS of 0.9974 (95% C.I., 17 0.9381-1.0567), 1.0366 (95% C.I., 0.9737-1.0995), 1.0333 (95% C.I., 0.9699-1.0967) and 1.0304 (95% C.I., 0.9669-1.0939) between the outgroup, GX_P1E, and the three ingroup strains. The largest value is observed for RaTG13 (1.0366), despite this sequence being the most early sampled sequence, perhaps caused by additional undetected recombination into RaTG13. Another possibility is that RaTG13 has been maintained under conditions which have allowed it to continue to evolve after its initial sampling.
Whole genome dN and dS estimates among the viral strains. The dS estimates are shaded in green, and the dN estimates are in orange shade. The 95% confidence intervals, calculated based on 1,000 bootstrap replicates, are included in the brackets for each estimates.
Genome-wide dN and dS estimates after removing recombination regions inferred by 3SEQ. The dS estimates are shaded in green, and the dN estimates are in orange shade. The coordinates relative to the Wuhan-Hu-1 genome of the masked region can be found in the method section. The 95% confidence intervals, calculated based on 1,000 bootstrap replicates, are included in the brackets for each estimates.
Sliding windows of synonymous divergence
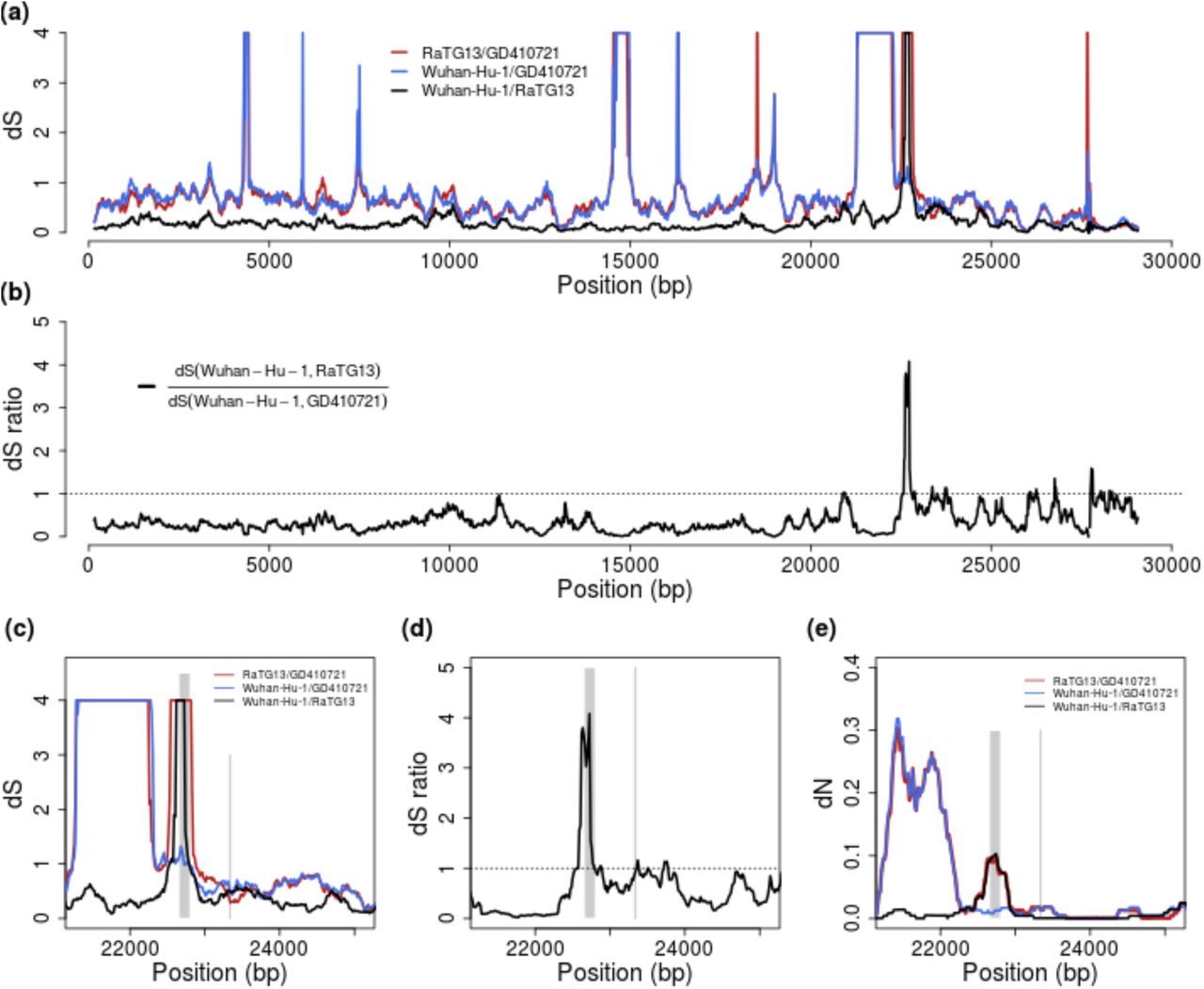
To address the issue of possible recombination we plot dS between SARS-CoV-2, GD410721, and RaTG13 and the ratio of dS(SARS-CoV-2, GD410721) to dS(SARS-CoV-2, RaTG13) in 300 bp sliding windows along the genome. Notice that we truncate the estimate of dS at 3.0. Differences between estimates larger than 2.0 should not be interpreted strongly, as these estimates have high variance and likely will be quite sensitive to the specifics of the model assumptions. We find that dS(SARS-CoV-2, GD410721) approximately equals dS(GD410721, RaTG13) and is larger than dS(SARS-CoV-2, RaTG13) in almost the entire genome showing than in these parts of the genome GD410721 is a proper outgroup to (SARS-CoV-2, RaTG13). One noticeable exception from this is the RBD region of the S gene. In this region the divergence between SARS-CoV-2 and GD410721 is substantially lower than between GD410721 and RaTG13 (Fig. 4A,C). The same region also has much smaller divergence between SARS-CoV-2 and GD410721 than between SARS-CoV-2 and RaTG13 (Fig. 4A,C). The pattern is quite different than that observed in the rest of the genome, most easily seen by considering the ratio of dS(SARS-CoV-2, GD410721) to dS(SARS-CoV-2, RaTG13) (Fig. 2B,D). In fact, the estimates of dS(SARS-CoV-2, RaTG13) are saturated in this region, even though they are substantially lower than 1 in the rest of the genome. This strongly suggests a recombination event in the region and provides independent evidence of that previously reported based on amino acid divergence (e.g., (Zhang, et al. 2020)).
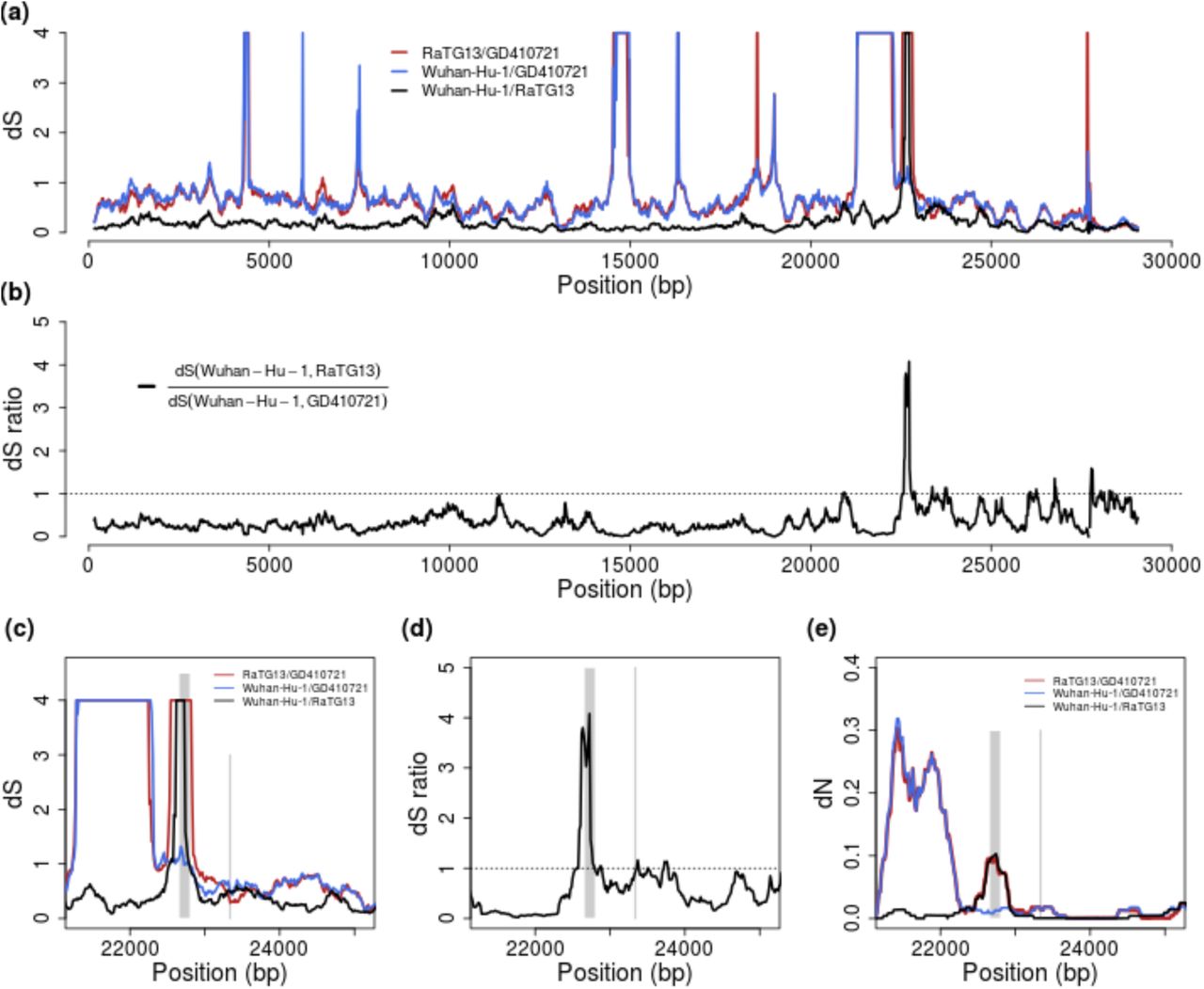
(a) Pairwise dS estimates in 300-bp sliding windows for RaTG13, GD410721 and Wuhan-Hu-1, the estimates are truncated at 4. (b) dS ratio of dS (Wuhan-Hu-1,RaTG13) to dS(Wuhan-Hu-1,GD410721). (c) and (d) are the zoom-in plot for dS and dS-ratio at the spike (S) protein region. The receptor-binding domain contact residues (left) and furin site regions (right) are marked with grey lines. (e) the pairwise dN estimates in 300-bp sliding windows in the S protein for these strains. The dS values are truncated at 4 in the plots.
The combined evidence from synonymous divergence and the topological recombination inference procedure in 3SEQ, provide strong support for the recombination hypothesis. However, these analyses alone do not distinguish between recombination into RaTG13 from an unknown source as hypothesized by Boni et al. (Boni, et al. 2020) and recombination between SARS-CoV-2 and GD410721 as proposed as one possible explanation by Lam et al. (Lam, et al. 2020). To distinguish between these hypotheses we searched for sequences that might be more closely related, in the RBD region, to RaTG13 than SARS-CoV-2 and we plotted sliding window similarities across the genome for RaTG13 (Fig. 1C). We observe relatively low sequence identity between RaTG13 and all three other strains in the ACE2 contact residue region of the spike protein, which is more consistent with the hypothesis of recombination into RaTG13, as proposed by Boni et al. (Boni, et al. 2020). Moreover, our BLAST search analyses of RaTG13 in this region show highest local sequence similarity with GX pangolin virus strains which is the genome-wide outgroup for the three other sequences (Lam, et al. 2020) (Lam, et al. 2020). This observation is more compatible with the hypothesis of recombination from a virus related to GX pangolin strains, than with recombination between SARS-CoV-2 and GD410721.
Unfortunately, because of the high level of synonymous divergence to the nearest outgroup, tree estimation in small windows is extremely labile in this region. In fact, synonymous divergence appears fully saturated in the comparison with GX_P1E, eliminating the possibility to infer meaningful trees based on synonymous divergence. However, we can use the overall maximum likelihood tree using both synonymous and nonsynonymous mutations (Fig. 2D). The ML tree using sequence from the ACE2 contact residue region supports the clustering of SARS- CoV-2 and GD410721, but with unusual long external branches for all strains except SARS- CoV-2, possibly reflecting smaller recombination regions within the ACE2 contact residue region.
Weakly deleterious mutations and clock calibrations
The use of synonymous mutations provides an opportunity to calibrate the molecular clock without relying on amino acid changing mutations that are more likely to be affected by selection. The rate of substitution of weakly and slightly deleterious mutations is highly dependent on ecological factors and the effective population size. Weakly deleterious mutations are more likely to be observed over small time scales than over long time scales, as they are unlikely to persist in the population for a long time and go to fixation. This will lead to a decreasing dN/dS ratio for longer evolutionary lineages. Furthermore, changes in effective population size will translate into changes in the rate of substitution of slightly deleterious mutations. Finally, changes in ecology (such as host shifts, host immune changes, changes in cell surface receptor, etc.) can lead to changes in the rate of amino acid substitution. For all of these reasons, the use of synonymous mutations, which are less likely to be the subject of selection than nonsynonymous mutations, are preferred in molecular clock calculations. For many viruses, the use of synonymous mutations to calibrate divergence times is not possible, as synonymous sites are fully saturated even at short divergence times. However, for the comparisons between SARS-CoV-2 and RaTG13, and SARS-CoV-2 and RmYN02, synonymous sites are not saturated and can be used for calibration. We find an estimate of ω = 0.0391 between SARS-CoV-2 and RaTG13, excluding just the small RDB region inferred by 3SEQ to be recombined in SARS-CoV-2 (Supplementary Table 5, coordinates: 22851-23094). Using 1000 parametric simulations under the estimated values and the F3×4 codon model, we find that the estimate is approximately unbiased  and with standard deviation 0.0033, providing an approximate 95% confidence interval of (0.0332, 0.0464). Also, using 59 human strains of SARS-CoV-2 from Genbank and National Microbiology Data Center (See Methods) we obtain an estimate of ω = 0.5604 using the F3×4 model in codeml. Notice that there is a 14-fold difference in dN/dS ratio between these estimates. Assuming very little of this difference is caused by positive selection, this suggests that the vast majority of mutations currently segregating in the SARS-CoV-2 are slightly or weakly deleterious for the virus.
and with standard deviation 0.0033, providing an approximate 95% confidence interval of (0.0332, 0.0464). Also, using 59 human strains of SARS-CoV-2 from Genbank and National Microbiology Data Center (See Methods) we obtain an estimate of ω = 0.5604 using the F3×4 model in codeml. Notice that there is a 14-fold difference in dN/dS ratio between these estimates. Assuming very little of this difference is caused by positive selection, this suggests that the vast majority of mutations currently segregating in the SARS-CoV-2 are slightly or weakly deleterious for the virus.
Dating of divergence between Bat viruses and SARS-CoV-2
To calibrate the clock we use the estimate provided by (http://virological.org/t/phylodynamic-analysis-of-sars-cov-2-update-2020-03-06/420) of μ =1.04×10−3 substitutions/site/year (95% CI: 0.71×10-3, 1.40×10-3). The synonymous specific mutation rate can be found from this as dS/year = μS = /(pS +ωpN), where pN and pS are the proportions of nonsynonymous and synonymous sites, respectively. The estimate of the total divergence on the two lineages is then  . Inserting the numbers from Table 3 for the divergence between SARS-CoV-2 and RaTG13 and RmYN02, respectively, we find a total divergence of 96.92 years and 74.05 years respectively. Taking into account that RaTG13 was isolated July 2013, we find an estimated tMRCA between that strain and SARS-CoV-2 of
. Inserting the numbers from Table 3 for the divergence between SARS-CoV-2 and RaTG13 and RmYN02, respectively, we find a total divergence of 96.92 years and 74.05 years respectively. Taking into account that RaTG13 was isolated July 2013, we find an estimated tMRCA between that strain and SARS-CoV-2 of  years. Similarly, we find an estimate of divergence between SARS-CoV-2 and RmYN02 of
years. Similarly, we find an estimate of divergence between SARS-CoV-2 and RmYN02 of  years, assuming approximately equal sampling times. The estimate for SARS-CoV-2 and RaTG13 is compatible with the values obtained by Boni et al. (Boni, et al. 2020), who used quite different methods for dating. The variance in the estimate in dS is small and the uncertainty is mostly dominated by the uncertainty in the estimate of the mutation rate. We estimate the S.D. in
years, assuming approximately equal sampling times. The estimate for SARS-CoV-2 and RaTG13 is compatible with the values obtained by Boni et al. (Boni, et al. 2020), who used quite different methods for dating. The variance in the estimate in dS is small and the uncertainty is mostly dominated by the uncertainty in the estimate of the mutation rate. We estimate the S.D. in  using 1000 parametric simulations, using the ML estimates of all parameters, for both RaTG13 vs. SARS-CoV-2 and for RmYN02 vs. SARS-CoV-2, and for each simulated data also simulating values of μ and ω from normal distributions with mean 1.04×10−3 and S.D. 0.18×10−3, and mean 0.5604 and S.D. 0.1122, respectively. We subject each simulated data set to the same inference procedure as done on the real data. Our estimate of the S.D. in the estimate is 11.8 for RaTG13 vs. SARS-CoV-2 and 9.41 for RmYN02 vs. SARS-CoV-2, providing an approximate 95% confidence interval of (28.11, 75.31) and (18.19, 55.85), respectively. For RaTG13, if including all sites, except the 244-bp in the RBD of the S gene (Supplementary Table 5), the estimate is 55.02 years with an approx. 95% C.I. of (29.4, 80.7). As more SARS-CoV-2 sequences are being obtained, providing more precise estimates of the mutation rate, this confidence interval will become narrower. However, we warn that the estimate is based on a molecular clock assumption and that violations of this assumption eventually will become a more likely source of error than the statistical uncertainty quantified in the calculation of the confidence intervals. We also note that, so far, we have assumed no variation in the mutation rate among synonymous sites. However, just from the analysis of the 300 bp windows, it is clear that is not true. The variance in the estimate of dS among 300 bp windows from the RaTG13-SARS-CoV-2 comparison is approximately 0.0113. In contrast, in the simulated data assuming constant mutation rate, the variance is approximately 0.0034, suggesting substantial variation in the synonymous mutation rate along the length of the genome. Alternatively, this might be explained by undetected recombination in the evolutionary history since the divergence of the strains.
using 1000 parametric simulations, using the ML estimates of all parameters, for both RaTG13 vs. SARS-CoV-2 and for RmYN02 vs. SARS-CoV-2, and for each simulated data also simulating values of μ and ω from normal distributions with mean 1.04×10−3 and S.D. 0.18×10−3, and mean 0.5604 and S.D. 0.1122, respectively. We subject each simulated data set to the same inference procedure as done on the real data. Our estimate of the S.D. in the estimate is 11.8 for RaTG13 vs. SARS-CoV-2 and 9.41 for RmYN02 vs. SARS-CoV-2, providing an approximate 95% confidence interval of (28.11, 75.31) and (18.19, 55.85), respectively. For RaTG13, if including all sites, except the 244-bp in the RBD of the S gene (Supplementary Table 5), the estimate is 55.02 years with an approx. 95% C.I. of (29.4, 80.7). As more SARS-CoV-2 sequences are being obtained, providing more precise estimates of the mutation rate, this confidence interval will become narrower. However, we warn that the estimate is based on a molecular clock assumption and that violations of this assumption eventually will become a more likely source of error than the statistical uncertainty quantified in the calculation of the confidence intervals. We also note that, so far, we have assumed no variation in the mutation rate among synonymous sites. However, just from the analysis of the 300 bp windows, it is clear that is not true. The variance in the estimate of dS among 300 bp windows from the RaTG13-SARS-CoV-2 comparison is approximately 0.0113. In contrast, in the simulated data assuming constant mutation rate, the variance is approximately 0.0034, suggesting substantial variation in the synonymous mutation rate along the length of the genome. Alternatively, this might be explained by undetected recombination in the evolutionary history since the divergence of the strains.
Discussion
The unusually skewed distribution of nucleotide frequencies in synonymous sites in SARS-CoV-(Kandeel, et al. 2020), along with high divergence, complicates the estimation of synonymous divergence in SARS-CoV-2 and related viruses. In particular, in the third codon position the nucleotide frequency of T is 43.5% while it is just 15.7% for C. This resulting codon usage is not optimized for mammalian cells (e.g, Chamary, et al. 2006). A possible explanation is a strong mutational bias caused by Apolipoprotein B mRNA-editing enzymes (APOBECs) which can cause Cytosine-to-Uracil changes (Giorgio, et al. 2020).
A consequence of the skewed nucleotide frequencies is a high degree of homoplasy in synonymous sites that challenges estimates of dS. We here evaluated estimators of dS in 300 bp sliding windows and found that a bias-corrected version of the maximum likelihood estimator tended to perform best for values of dS < 2. We used this estimator to investigate the relationship between SARS-CoV-2 and related viruses in sliding windows. We show that synonymous mutations show shorter divergence to pangolin viruses, than the otherwise most closely related bat virus, RaTG13, in part of the receptor-binding domain of the spike protein. This strongly suggests that the previously reported amino acid similarity between pangolin viruses and SARS-CoV-2 is not due to convergent evolution, but more likely is due to recombination. Boni, et al. 2020 used the program 3SEQ to infer recombination events in SARS-CoV-2 and related viruses and did not recover strong evidence for recombination specific to SARS-CoV-2. In contrast in our analyses, when applying the program to a smaller subset of viral strains, 3SEQ identifies recombination from pangolin strains into SARS-CoV-2. Possibly, the inclusion of more divergent strains in the more comprehensive analysis by (Boni, et al. 2020) has lead to reduced power to detect recombination among the less divergent strains closely related to SARS-CoV-2. In any case, the 3SEQ analyses presented provides further support for the recombination hypothesis. However, we also find that the synonymous divergence between SARS-CoV-2 and pangolin viruses in this region is relatively high, suggesting that the recombination was into RaTG13 from an unknown strain, rather than between pangolin viruses and SARS-CoV-2, which is consistent with the hypothesis proposed by Boni, et al. 2020, and we can exclude the possibility of a very recent recombination event between pangolins and SARS-CoV-2. Another alternative explanation is that there is a mutational hotspot located in the RBD region. While it is possible that selection may have favored hypermutability in this domain, the most parsimonious explanation at the moment, in the absence of knowledge of molecular mechanisms to explain such a hotspot, is recombination into the RaTG13 strain. To fully distinguish between these hypotheses, additional strains would have to be discovered that either are candidates for introgression into RaTG13 or can break up the lineage in the phylogenetic tree between pangolin viruses and RaTG13.
The fact that synonymous divergence to the outgroups, RaTG13 and RmYN02, is not fully saturated, provides an opportunity for a number of different analyses. First, we can date the time of the divergence between the bat viruses and SARS-CoV-2 using synonymous mutations alone. In doing so, we find estimates of 51.71 years (95% C.I., 28.11-75.31) and 37.02 years (95% C.I., 18.19-55.85), respectively. Most of the uncertainty in these estimates comes from uncertainty in the estimate of the mutation rate reported for SARS-CoV-2. As more data is being produced for SARS-CoV-2, the estimate should become more precise and the confidence interval significantly narrowed. However, we warn that a residual cause of unmodeled statistical uncertainty is deviations from the molecular clock. Variation in the molecular clock could be modeled statistically (see e.g., Drummond, et al. 2006 and Lartillot, et al. 2016), but the fact that synonymous mutations are mostly saturated for more divergent viruses that would be needed to train such models, is a challenge to such efforts. On the positive side, we note that the estimates of dS given in Table 3 in general are highly compatible with a constant molecular clock.
Another advantage of estimation of synonymous and nonsynonymous rates in the outgroup lineage, is that it can provide estimates of the mutational load of the current pandemic. The dN/dS ratio is almost 14 times larger in the circulating SARS-CoV-2 strains than in the outgroup lineage. While some of this difference could possibly be explained by positive selection acting at a higher rate after zoonotic transfer, it is perhaps more likely that a substantial proportion of segregating nonsynonymous mutations are deleterious, suggesting a very high and increasing mutation load in circulating SARS-CoV-2 strains.
Acknowledgements
We are grateful to Dr. Yongyi Shen and Dr. E.C Holmes for providing the genome sequence ofGD410721 and RmYN02, respectively. We also thank Dr. Adi Stern for discussion. The research was funded by Koret-UC Berkeley-Tel Aviv University Initiative in Computational Biology and Bioinformatics to RN.





{kind=link}
{kind=link}
{kind=link}
{kind=link}